6.2.3.1 酶与底物的作用学说
(1) 锁钥学说 1894年,E.Fisher提出酶与底物作用的锁钥学说(lock and key theory),用以解释酶作用的专一性。这个学说认为,酶与底物分子或底物分子的一部分之间, 在结构上有严格的互补关系。当底物契合到酶蛋白的活性中心时,很像一把钥匙插入到 一把锁中。酶的天然构象是刚性的,如果底物分子结构上存在着微小的差别,就不能契入 酶分子中,从而不能被酶作用。
利用“锁钥学说”再结合“酶与底物的三点附着学说”,对于了解酶的立体化学专一性、 酶按次序地和两个或更多个底物结合以及简单的底物饱和曲线动力学,均有所帮助。但 还有些问题是这些理论所不能解释的,如果酶的活性中心是“锁钥学说”中的锁,那么这种 结构不可能既适合于可逆反应的底物,又适合于可逆反应的产物。并且,上述理论也不可 能解释酶的专一性中的所有现象。
(2)诱导契合学说 上述学说对很多酶来说并不全面,越来越多的事实说明“锁钥学 说”虽然有合理的内核,但与许多实验事实不符。1958年,Koshland提出了“诱导契合学 说”(induced-fithypothesis),其主要意思是:当酶分子与底物分子接近时,酶蛋白受底物 分子的诱导,其构象发生有利于底物结合或催化的变化。酶与底物在这个基础上互补契 合,进行反应。
“诱导契合学说”不仅能解释“锁钥学说”不能解释的实验事实,而且近年来运用各种 物理化学方法,如X-射线衍射分析、旋光色散、圆二色性、核磁共振、差示光谱等,确实证 实了酶与底物结合时,酶的构象发生了变化。
6.2.3.2 酶的动力学学说
酶动力学主要是研究各种因素对酶催化反应速度的影响,这些因素包括底物浓度、酶 浓度、产物、pH、温度、抑制剂和激活剂等。研究酶动力学对了解酶的作用机制、选择合适 的生产工艺及酶反应器的设计皆有重要的意义。
(1) 单底物动力学
①Michaelis-Menten学说: 1903年,Henri在研究蔗糖酶水解蔗糖的反应中发现, 在恒定的温度、pH及酶浓度条件下,底物浓度对酶的催化反应速度产生影响。当底物浓 度很低时,反应速度随底物浓度的增大而迅速增快。而后底物浓度继续增大时,反应速度 的增率就比较小。当底物浓度增大至某种程度时,反应速度达到一个极限值。此后如果 再增大底物浓度,反应速度保持此值不再增快,故酶催化反应的速度与底物浓度呈现双曲 线关系。据此,Henri提出,酶和底物的作用是通过酶和底物生成复合物而进行的。当底 物浓度较低时,酶的活力中心未被饱和,因此,反应速度随底物浓度上升而呈正相关。当 底物浓度较高,即酶的活性中心被饱和或趋于饱和时,反应速度增率较小或不再增快。这 是因为酶-底物复合物的生成速度相对较快,而分解速度相对较慢,成为整个反应的限速 步骤。这就是中间复合物学说,可以表达为:

(6-1)
式中 E——游离酶
S——底物
ES——酶-底物复合物
P——产物
k-1/k1——ES的解离常数,以Ks表示
k2——ES复合物分解的反应速度常数
1913年,Michaelis和Menten根据中间复合物学说,对式(6-1)进行了数学处理,其 推导过程基于以下三点假说:
a. 假设式(6-1)中没有考虑E+P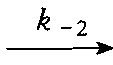 ES的逆反应,对大多数酶而言,k-2是一个 不等于零的常数,要忽略这一步,必须是P→0。只有测定的速度是反应的初速度,此时S 的消耗很小(消耗量在5%以内),P的生成极少,才能忽略。
ES的逆反应,对大多数酶而言,k-2是一个 不等于零的常数,要忽略这一步,必须是P→0。只有测定的速度是反应的初速度,此时S 的消耗很小(消耗量在5%以内),P的生成极少,才能忽略。
b. 假设底物浓度是以起始浓度[S0]计算的,这就要求底物浓度大大超过酶的相对 浓度,否则由于ES的存在,[S]就不能用[S0]代替。
c. 假设ES解离成E+S的速度显著快于ES形成P+E的速度,即k-1>k2,或者说 E+S⇌ES的可逆反应在初速度测定的时间内就已达到平衡。 ES分解生成产物的速 度不足以破坏E和S之间的平衡,这就是“平衡学说”。
基于上述三点假设得出速度方程:

(6-2)
这里 KS=k-1/k1
式中 vmax——最大反应速度
v——反应初速度
这就是Michaelis-Menten方程,简称米氏方程。它表达了反应初速度与底物浓度的 关系。如果作vmax~[S]图,是一条矩形双曲线。式(6-2)长期以来与大量实验结果基 本符合,已经广为人们所接受。
②Briggs-Haldane修正公式: 1925年,Briggs和Haldane对米氏方程作了一项重要 修正。他们认为,当k2>k-1时,Michaelis和Menten的假设中的快速平衡不一定能够成 立,就不能用上述“平衡学说”予以推导。他们考虑到比式(6-1)更有普遍性的反应式:

(6-3)
并提出了稳态(steady-state)学说。这个学说认为:在初速度的测定过程中,[S]随时间 降低,[P]相应增高,而中间复合物ES在一开始增高后,可在相当一段时间内保持浓度的 恒定。在这段时间内,ES生成的速度和ES消失的速度(包括形成E+S和E+P)相等, 达到动态平衡,这就是所谓的“稳态”。在公式推导时,他们保留了Michaelis和Menten的 前面两点假设,而将第三点“平衡”假设修改成“稳态”假设。
根据Briggs和Haldane的修正,推导得速度方程:

(6-4)
式中,Km=(k2+k-1)/k1称米氏常数。
式(6-4)和Michaelis-Menten推导的方程有相同的形式,但Km的含义与Ks不 同,具有更大的普遍性。当k2<<k-1时,

(6-5)
为了纪念Michaelis和Menten的开创性的工作,习惯上把式(6-2)和式(6-4)、式 (6-5)都称为Michaelis-Menten方程。
③关于Michaelis-Kenten方程的讨论
a. 根据米氏方程,酶反应的速度和底物浓度的关系为一双曲线。当[S]>>Km时,式 (6-4)简化为v=vmax,成为零级反应速度方程。在测定酶活力时,一般选择底物饱和或 者零级动力学的条件。在这样的条件下,酶活力直接正比于酶浓度而与底物浓度无关,当 [S]<<Km时,式(6-4)简化为:

(6-6)
成为一级反应速度方程,或v=kapp[S],此处kapp=vmax/Km。这个原理在酶法分析中被 应用。利用酶测定底物时,可使用足够量的酶以便在较短的时间内,使反应达到完全。这 样测定形成的产物总量就和待测物的量相等或相关。
Km是反应底物浓度与反应速度关系的动力学常数。从米氏方程可以直接推出,Km 值等于达到最大反应速度一半时的底物浓度。对某一个酶相应的底物来说,Km是其特 征性常数。当pH、温度、离子强度及其他有关因素不变时,Km应是恒定的。纯度不同的 同一种酶,如果不含有引起底物其他变化的杂质时,其Km值也不变。
b. 因为vmax=k2[E],当v=vmax时,反应初速度与底物浓度无关,只和[E]成正比。 此时酶的活性中心被饱和,当Km已知时,任何底物浓度对酶活性中心被底物饱和的份数 (saturation fraction,fes)可以用下式表示:

(6-7)
c. 由式(6-4)可知,当k2<<k-1时,Km=k-1/k1=Ks,也即Km等于ES复合物的解 离常数,可以作为酶和底物结合紧密程度的一个量度。但在不知道Km确实等于Ks之 前,用Km表示酶和底物的亲和力是不行的。
d. 式(6-2)和式(6-4)中,vmax代替k2[E],k2为一级反应速度常数,其量纲为 s-1,称为酶的转换系数(turnover number,T.N.)。T.N.越大,表示酶的催化效率越高。 T.N.的求取可由V/[E]计算,式中[E]用活性部位的当量表示。
在实际应用过程中,一个酶的米氏常数对其性能是可以做出某些暗示的。米氏常数 是一个酶的特征性常数,其值等于在一定的实验条件下达到1/2V时的底物浓度。因此, 它给出一个酶能够应答底物浓度变化范围的有用指示。各种酶的米氏常数不同,同一种 酶对不同底物的米氏常数也不同。如果能够证明ES复合体分解形成产物的速率要比它 从底物形成的速率慢得多,即k-1远大于k2的话,Km是可以看做ES的解离常数的,就 可以用于表示酶与底物亲和力的大小。在这种情况下,米氏常数越小,表示酶与底物的亲 和力越大,在较低的底物浓度时已经能使反应速度达到相对较高。如果一个酶能作用于 几个底物,那么具有最小Km的底物通常可能是“天然”的底物。否则,比较不同来源的酶 对同一底物的Km值,也可以帮助了解它们是否有差别。
(2) 双底物动力学 上节所讨论的动力学只适用于单底物的反应,而大多数酶催化 的反应是两个或多个底物间的反应。按照EC规定分成的六大类酶中,真正单底物的反 应只有异构酶类和裂解酶类。水解酶类催化的反应被认为是假单底物反应,其动力学往 往符合单底物反应的动力学,因为水在反应过程中变化甚微,可以作为常量处理。其余酶 类催化的反应皆为双底物或多底物反应,其中以转移反应占绝大多数。氧化还原反应是 转移电子或氢原子的反应,也可以看作是转移反应的一种。这里讨论的多底物动力学也 以转移反应作为模式。
多底物动力学的机制分为两大类,一类是序列(Sequential)反应,另一类是乒乓(Ping Pong)反应。下面以双底物反应物系统为例分别介绍这两类机制的特征。
①序列机制: 其主要特征为底物的结合和产物的释放有一定顺序。产物不能在底 物完全被结合以前释放。序列反应又分为有序反应(Ordered)和随机反应(Random)。
a. 序列有序反应: 在这个机制中,底物A与酶结合后,酶的构象发生变化,暴露出与 底物B的结合部位,使B与酶结合,用图解表示为:

图中:A、B、C…分别表示按先后与酶结合的底物;P、Q、R…分别表示按先后释放的产物; E、F、G…代表酶的游离形式,也是酶的稳定形式;(EAB)、(EPQ)等表示酶被底物饱和的 中心复合物;(EAB-EPQ)或(EA-EP)表示其异构作用。以下同样表示。
b. 序列随机反应: 其主要特征为两个底物A和B能随机地与酶结合,其产物P和 Q也可以随机地脱离酶,用图解可表示为:

②乒乓机制: 其特征为酶E与底物A生成复合物,产物P的脱离在另一底物B加 入之前,用图解可以表示为:

酶促多底物反应机制的鉴别,可以通过底物动力学作图,还可以通过同位素交换法和 产物抑制动力学法进行鉴别。同位素交换法不但可以区别序列机制和乒乓机制,还可以 区别序列机制中的有序机制和随机机制。
6.2.3.3 影响酶反应动力学的因素
(1) 酶浓度 在酶反应中,如果底物浓度足以使酶饱和,则反应速度与酶浓度成正 比。这种关系也可以由米氏方程推导得出:
 又 v=k2[E]
又 v=k2[E]
所以:

当[S]维持不变时,v∝[E]。这里使用的酶必须是纯酶制剂或不含抑制剂的粗酶制剂。
(2) pH 大部分酶的活力受其环境pH的影响。在一定的pH条件下,酶反应具有 最大速度。高于或低于此值,反应速度下降,通常称此pH为酶反应的最适pH。用酶活 力对pH作图,往往得到钟罩形的曲线。酶的最适pH目前还只能用实验方法得到,它可 以随着底物的浓度、温度和其他条件的变化而变化。因此,酶的最适pH并不是一个常 数,它只是在一定的条件下才有意义。
pH对酶活力的影响主要表现在以下几个方面:
①pH的改变可以破坏酶的空间构象,引起酶活力的丧失。这种失活作用或者是可 逆的,或者是不可逆的。
②pH的改变影响酶的活性中心催化基团的解离,从而使得底物转变成产物的过程 受到影响。
③pH的改变影响酶的活性中心结合基团的解离状态,使得底物不能与其结合。
④pH的改变影响底物的解离状态,或者使底物不能和酶结合,或者结合后不能生成 产物。
pH可以对游离酶或酶-底物复合物产生一定的效应,从而导致对酶反应的速度产生 显著的影响,其机理是相当复杂的。
(3) 温度 温度对酶反应速度也有很大的影响,温度的影响通常包括两个方面:一方 面是当温度升高时,与一般化学反应一样,反应速度加快;另一方面,随着温度升高而使酶 蛋白逐步变性,反应速度随之下降。因此,酶反应存在着一个最适温度(optimum temperature)。如果以反应速度对温度作图,所得的曲线为钟罩形,曲线顶点即为最适温度,最适 温度是上述两个方面平衡的结果。最适温度不是酶的特征性物理常数,它往往受到作用 时间、酶的浓度、底物、激活剂或抑制剂等因素的影响。因此,对某一种酶而言,必须说明 是在什么条件下的最适温度。
在变性温度以内,温度对酶催化反应速度的影响也服从Arrhenius方程。温度每增高 10℃,酶催化反应速度增快1~2倍。
(4) 抑制剂 能降低酶催化反应速度的因素很多,通常将其分为失活作用和抑制作 用两类。失活作用是指由于一些物理因素和化学试剂部分或全部破坏了酶的三维结构, 即引起酶蛋白变性,导致其部分或全部丧失活力。抑制作用是指在酶不变性的情况下,由 于必需基团或活性中心化学性质的改变而引起的酶活力的降低或丧失。导致抑制作用产 生的物质称为酶抑制剂。
①不可逆抑制作用: 导致这类抑制作用的抑制剂,通常以比较牢固的共价键与酶蛋白中 的基团结合,从而使酶失活。失活的酶不能用透析、过滤等物理方法除去抑制剂而恢复酶活力。
根据不可逆抑制作用的选择性,可分为非专一性的不可逆抑制和专一性的不可逆抑制。
非专一性的不可逆抑制剂的作用是不专一的,它不但和酶的必需基团作用,同时也和 酶的非必需基团作用,甚至还会和不止一种类型的侧链基团作用。这类抑制剂主要是一 些修饰氨基酸侧链基团的化学试剂,可与氨基、巯基、羟基、胍基、酚基等反应。
专一性不可逆抑制剂仅仅和活性中心有关的基团反应。属于这一类的抑制剂又可分 为Ks型和Kcat型两类。Ks型不可逆抑制剂是根据底物的结构设计的,它具有和底物相似 的结合基团,同时还具有一个能和活性中心的其他功能基团反应的活性基团。对于Ks型 抑制剂,通常需要判断抑制作用是否发生在活性部位。Kcat型不可逆抑制剂是根据酶的 催化过程设计的。它不但具有与天然底物类似的被酶结合和被酶催化的反应性质,此外 还有一个潜伏的反应基团,这个基团可因酶的催化而暴露或活化,并作用于酶活性中心基 团或酶的辅酶,导致酶被共价修饰而失活。这类抑制剂的专一性很高。
②可逆抑制作用: 导致这类抑制作用的抑制剂与酶蛋白的结合通常是可逆的,可用 透析等物理方法除去抑制剂,恢复酶的活力。根据抑制剂与底物的关系,可逆抑制作用分 为四种类型。
a. 竞争性抑制作用: 这是比较常见的可逆抑制作用,常常是由于抑制剂与底物在结 构上有类似之处,因此抑制剂(I)和底物(S)互相竞争地与游离酶E结合。已结合S的ES 复合物不能再结合I;已结合I的EI复合物也不能再结合S,即不可能存在EIS三联复合 物。反应式可表示为:

推导得到其动力学方程为:

b. 反竞争抑制作用:这类抑制剂I不和游离酶结合,只能和ES中间复合物结合成 EIS,但EIS不能释出产物。S和E的结合不但不排斥I,反而促进了I和E的结合,可表示如下:

其动力学方程为:

c. 非竞争性抑制作用: 游离酶可以和这类抑制剂I结合,生成EI,也可以和底物S 结合,生成ES。酶和底物的结合并不影响酶和抑制剂的结合,可以生成EIS,但EIS不能 释放出产物。可表达如下:

推导可得其动力学方程为:

d. 线性混合型抑制作用: 混合型抑制的机制基本上与非竞争性抑制作用相似,S或 I和E的结合不相关,ES可再结合I,EI也可以再结合S,但Ks不等于Ks′,Ki也不等于 Ki′,可以表示如下:

推导得其动力学方程为:

上述四种抑制类型在实验上是比较容易区分的,其抑制程度与底物浓度的关系显然 各不相同:
(a)底物浓度的改变对抑制程度没有影响,这是非竞争性抑制的特征。
(b)底物浓度增大,抑制程度降低,这是竞争性抑制的特征。
(c) 底物浓度增大,抑制程度也增强,这是反竞争性抑制的特征。
③底物抑制和产物抑制
a. 底物抑制: 在酶反应中,有时会出现高浓度底物反而使反应速度下降的现象,称 为底物抑制。底物抑制作用在单底物和多底物反应中均存在。
底物抑制的产生机制,可能是[S]太高时,与ES中间复合物形成ES2,后者没有活 力,也不释放出产物,如下式表示:

当E恒定而S从零逐渐升高时,因ES不断增多,生成的ES2较少,反应速度逐渐上升至 最高值。此时如果再加大S,则ES2的生成逐渐增多,ES相应减少,故反应速度逐渐降低。
b. 产物抑制: 产物对酶反应的抑制是比较常见的。例如葡萄糖-6-磷酸对己糖激 酶,ATP对丙酮酸激酶的抑制,皆属此类。在酶反应中,产物在释出之前可以以酶-复合物 (EP)的形式存在。也就是说,产物也可以占领酶分子上的特殊位点,导致酶的催化速度下 降。在双底物、双产物反应中,产物还可以和催化过程中生成的不同的酶分子形式(如E、F、EP 或EQ等)结合,或者和酶-底物复合物结合,形成无效的死端产物,从而导致抑制效应的产生。
(5)激活剂 激活剂是实质能加快某种酶反应速度的物质,它们的作用与抑制剂相 反。其中大部分是离子或简单的有机化合物。离子的激活作用可能有以下三种机理:
①与酶分子肽链上的侧链基团相结合,稳定酶催化作用所需的构型。
②作为底物(或辅酶)与酶蛋白之间联系的桥梁。
③协助酶的催化作用。
一般地说,这三种功能是相互协同的。
简单的有机化合物对酶的激活,可能通过与游离酶相结合形成活性酶复合物,或者与 底物结合形成复合的活性底物,或者和酶-底物复合物形成三元络合物等,从而对酶起激 活作用,激活作用的机制和抑制作用类似。